Wound Healing
Wound healing is a highly coordinated biological process involving overlapping phases of cellular and molecular activity. Understanding these phases — and the factors that disrupt them — is fundamental to surgical planning, reconstructive timing, and complication avoidance. The process is classically divided into four phases: hemostasis, inflammation, proliferation, and remodeling.[1][2]
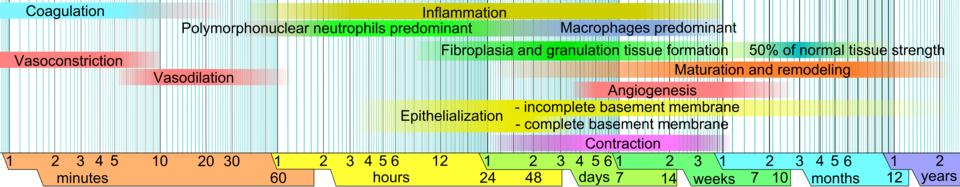
The four phases of wound healing — hemostasis, inflammation, proliferation, and remodeling — are temporally overlapping, not strictly sequential. Limits vary by wound size and healing conditions. (Public domain)
A wound achieves only ~80% of pre-injury tensile strength by approximately 60 days. Full remodeling continues for more than one year. This has direct implications for catheter removal timing, graft maturation, and re-operation planning.
Phase 1: Hemostasis / Clot Formation
Begins immediately at time of injury
The first response to tissue injury is cessation of bleeding through a coordinated vascular and platelet response:[1][3]
- Vasoconstriction: Immediate reflex vasoconstriction augmented by the release of thromboxane A2 and prostaglandins from injured cells reduces blood flow to the wound
- Platelet activation: Exposed subendothelial collagen activates platelets via von Willebrand factor (vWF) and platelet-activating factor; activated platelets release thromboxane A2, further amplifying vasoconstriction and platelet recruitment
- Platelet plug formation: Primary hemostasis — aggregated platelets form a loose mechanical plug
- Coagulation cascade: Fibrinogen is converted to fibrin (via thrombin), creating a fibrin mesh that reinforces the platelet plug and forms the provisional matrix — a scaffold for subsequent cellular migration
The fibrin clot provides weak initial wound tensile strength and serves as a reservoir of growth factors (PDGF, TGF-β, VEGF) released from platelet alpha-granules that initiate the subsequent inflammatory phase.
Phase 2: Inflammation
Begins immediately; dominant over first 72 hours
Inflammation serves to clear debris and bacteria while recruiting the cells necessary for repair:[1][2]
- Vasodilation: Histamine, serotonin, and bradykinin released from mast cells and platelets cause vasodilation and increased vascular permeability; endothelial cells swell, allowing extravasation of plasma and leukocytes
- Neutrophil influx (0–48 hours): Polymorphonuclear neutrophils (PMNs) are the first responders — they phagocytose bacteria and devitalized tissue, releasing proteases and reactive oxygen species. Neutrophils are the dominant cell type in the first 24–48 hours
- Macrophage dominance (48 hours onward): Monocytes recruited from blood differentiate into macrophages; these become the dominant cells after 48 hours and are the central orchestrators of the entire healing response. Macrophages phagocytose bacteria, dead PMNs, and debris, and release growth factors (PDGF, TGF-β, FGF, VEGF) that trigger proliferation
- Lymphocyte recruitment (late inflammation): T-lymphocytes enter the wound under the influence of interleukin-1, secreting cytokines that modulate macrophage activity and early fibroblast function
- Cell proliferation signal: Platelet-derived growth factor (PDGF) and other pro-inflammatory mediators released during this phase (serotonin, bradykinin, histamine) initiate cell proliferation and transition to the next phase
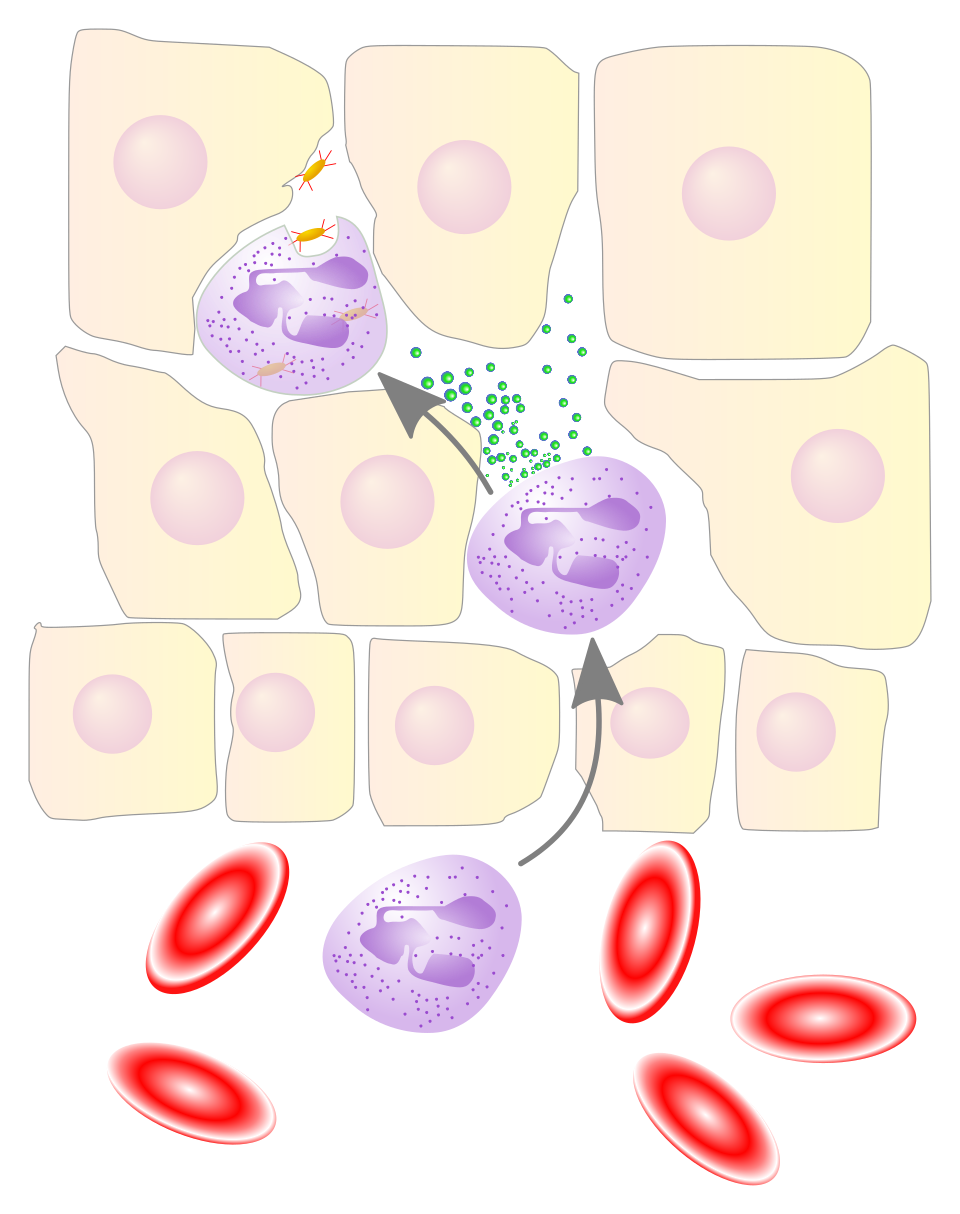
Neutrophil granulocytes migrate from the bloodstream into the wound matrix, secreting proteolytic enzymes and phagocytosing bacteria. They are the dominant cell type for the first 24–48 hours before macrophages take over. (CC0)
Cellular Sequence.
Neutrophils (0–48h) → Macrophages (dominant from 48h) → Lymphocytes (late inflammation / transition)
Macrophage depletion experiments demonstrate that wounds cannot progress to the proliferative phase without macrophages — they are indispensable orchestrators, not merely debris clearers.[4]
Phase 3: Proliferation
Begins ~72 hours after injury; complete within 2–4 weeks
The proliferative phase rebuilds the wound matrix and restores structural integrity:[1][2]
- Fibroblast migration and proliferation: Stimulated by PDGF and TGF-β, fibroblasts migrate into the provisional fibrin matrix and begin synthesizing extracellular matrix. Fibroblasts are the dominant cell type of this phase
- Matrix formation: Fibroblasts produce glycosaminoglycans (hyaluronic acid, chondroitin sulfate) and proteoglycans that provide hydration and structural organization to the new matrix
- Collagen synthesis: Fibroblasts predominantly synthesize type III collagen (fetal-type, thinner, less organized) — this provides significant but imperfect tensile strength. Type III predominates during proliferation; it is later replaced by type I during remodeling
- Wound contraction: Fibroblasts differentiate into myofibroblasts (under TGF-β influence), which contain smooth muscle actin and actively contract the wound margins. This reduces wound area but can lead to contracture deformity if excessive or in unfavorable locations
- Angiogenesis: Endothelial progenitor cells migrate and proliferate in response to VEGF (released by macrophages and hypoxic tissue); new capillary networks form, giving proliferating wounds their characteristic red, granular appearance
- Epithelialization: Keratinocytes at wound margins and adnexal structures migrate across the wound surface under a moist environment
Tensile Strength During Proliferation. By day 5, tensile strength begins to increase measurably as collagen cross-linking begins. However, the wound remains fragile — this is the rationale for protecting catheterized urethral repairs and graft recipient sites during the early weeks of healing.
Phase 4: Remodeling
Begins 2–4 weeks after injury; continues for >1 year
Remodeling reorganizes and matures the collagen matrix, ultimately determining the quality and appearance of the final scar:[1][2]
- Collagen homeostasis: Net collagen production ceases; a balance between synthesis and degradation (via matrix metalloproteinases, MMPs) is reached
- Type III → Type I collagen replacement: The disorganized type III collagen of early repair is progressively replaced by type I collagen (thicker, more organized, stronger). This conversion is the primary driver of increasing tensile strength during remodeling
- Collagen reorganization: Collagen fibrils align along lines of mechanical stress and form stronger intermolecular cross-links via lysyl oxidase
- Apoptosis and regression: The capillary network established during angiogenesis undergoes programmed apoptosis, reducing vascularity and creating the characteristic pale, avascular appearance of mature scar
- Tensile strength evolution: Tensile strength reaches ~80% of pre-injury strength by ~60 days and plateaus. Full pre-injury strength is never completely recovered
| Time | Tensile Strength | Dominant Process |
|---|---|---|
| Day 0–5 | Very low (clot only) | Hemostasis + early inflammation |
| Day 5–21 | Rapidly increasing | Collagen synthesis (Type III) |
| Day 21–60 | ~80% of normal | Type III → Type I conversion |
| 60 days – 1 year | Stable (~80%) | Cross-linking, remodeling |
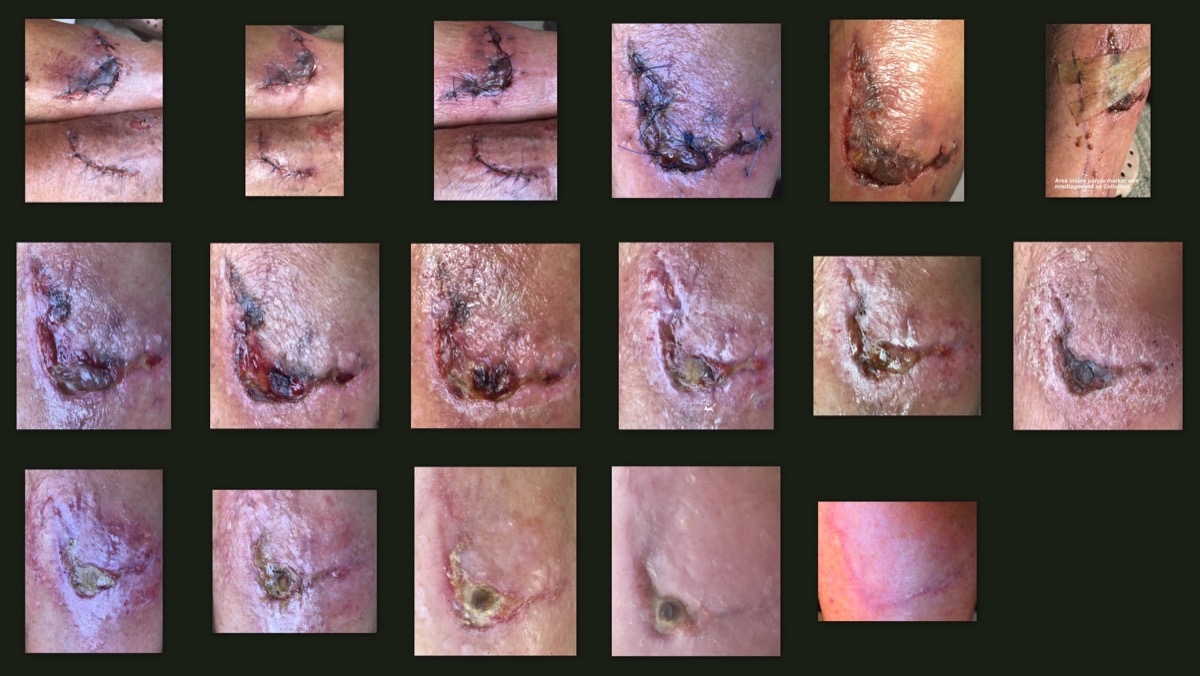
Serial photographs of a healing skin abrasion over 30 days, illustrating the visible progression through the inflammatory (erythema, swelling), proliferative (granulation, new epithelium), and early remodeling (fading, scar maturation) phases. (CC BY-SA 4.0)
Chronic Wound. In a chronic wound, healing stalls — most commonly in the inflammatory phase. Excessive matrix metalloprotease (MMP) activity promotes matrix degradation faster than synthesis. Bacterial biofilm, persistent ischemia, and repetitive trauma are common perpetuating factors. Management targets MMP suppression, infection control, and wound bed optimization.
Systemic Factors Affecting Wound Healing
| Factor | Mechanism of Impairment |
|---|---|
| Age | Decreased cellular proliferation, reduced growth factor signaling, impaired angiogenesis, reduced collagen synthesis |
| Smoking | Nicotine → vasoconstriction; carbon monoxide → reduced O₂-carrying capacity; hydrogen cyanide → impaired mitochondrial oxygen transport; net effect: tissue ischemia[5] |
| Diabetes mellitus | Impaired leukocyte function, reduced angiogenesis (decreased VEGF response), neuropathy, microvascular disease, elevated glucose impairs fibroblast proliferation |
| Malnutrition | Vitamin A (retinol) required for collagen cross-linking and epithelialization; Vitamin C required for collagen synthesis (hydroxylation of proline/lysine); low albumin is an independent predictor of poor wound healing[2] — see Perioperative Nutrition for the wound-healing evidence (200k-pt albumin data, protein RCTs, multinutrient bundles, GLP-1 RA management). |
| Corticosteroids | Inhibit epithelialization and fibroblast proliferation; decrease collagen production; impair macrophage function; dose-dependent effect |
| Immunosuppression | Impairs neutrophil and macrophage function; increases infection risk; delays transition from inflammation to proliferation |
| Radiation | Occludes small vessels; damages fibroblasts and DNA; creates permanently hypovascular, hypoxic tissue bed that heals poorly and is at high risk for wound breakdown after subsequent surgery[6] |
Local Wound Factors
Local factors are the most common cause of delayed or failed wound healing in surgical patients:[2]
| Factor | Effect |
|---|---|
| Infection | Reduces tissue oxygen tension and pH; impairs epithelialization and angiogenesis; increases collagenase activity; prolongs inflammation |
| Ischemia / Oxygen delivery | Microvascular disease → reduced tissue perfusion; hypoxia impairs fibroblast function, collagen synthesis, and leukocyte bactericidal activity |
| Radiation | Permanently damaged microvasculature; fibroblast dysfunction; reduced regenerative capacity; hallmark of post-radiation reconstructive challenge |
| Temperature | Warm, moist environment promotes epithelialization and tensile strength; cold or desiccated wounds heal more slowly |
| Wound tension | Excessive tension at wound edges impairs perfusion and increases dehiscence risk; a fundamental principle of plastic surgery is closure without tension |
| Foreign body / Suture material | Perpetuates inflammation; prolongs inflammatory phase; increases infection risk |
Video Resource
For a visual overview of the wound healing cascade:
Wound Healing — Mechanism and Phases (YouTube)
References
1. Diegelmann RF, Evans MC. Wound healing: An overview of acute, fibrotic and delayed healing. Front Biosci. 2004;9:283–289. PMID 14766366
2. Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453(7193):314–321. PMID 18480812
3. Singer AJ, Clark RAF. Cutaneous wound healing. N Engl J Med. 1999;341(10):738–746. PMID 10471461
4. Leibovich SJ, Ross R. The role of the macrophage in wound repair: A study with hydrocortisone and antimacrophage serum. Am J Pathol. 1975;78(1):71–100. PMID 1109560
5. Sørensen LT. Wound healing and infection in surgery: The clinical impact of smoking and smoking cessation. Arch Surg. 2012;147(4):373–383. PMID 22143127
6. Dormand EL, Banwell PE, Goodacre TEE. Radiotherapy and wound healing. Int Wound J. 2005;2(2):112–127. PMID 16722862